
The study of rare genetic diseases can be a valuable way of acquiring new biological insights A new paper from joint first authors Ye Zhu and Motoki Fujimaki investigates the mechanisms by which mutations lead to pathological changes in the disease β-Propeller protein-associated neurodegeneration (BPAN). This rare X-linked dominant disease is one of several conditions that manifest with neurodegeneration and brain iron accumulation. The gene that is mutated leading to loss of function in BPAN is the WD repeat domain 45 (WDR45) gene, which encodes the protein WIPI4. Given that WIPI4 is involved in autophagy, it has been assumed that BPAN pathology is primarily due to incapacitation of this intracellular clearance pathway.
However, in this Nature Cell Biology paper, the data indicate that WIPI4 depletion causes ferroptosis—a type of cell death induced by lipid peroxidation that is implicated in many neurodegenerative diseases, including Alzheimer’s, Parkinson’s, Huntington’s disease and motor neuron disease WIPI4 depletion increases localization of its interacting protein ATG2A at endoplasmic reticulum–mitochondrial contact sites, which enhances phosphatidylserine import into mitochondria. This results in increased mitochondrial synthesis of phosphatidylethanolamine, a major lipid prone to peroxidation, thus enabling ferroptosis. This mechanism has minimal overlap with classical ferroptosis stimuli but provides insights into the causes of neurodegeneration in BPAN and may provide clues for therapeutic strategies.
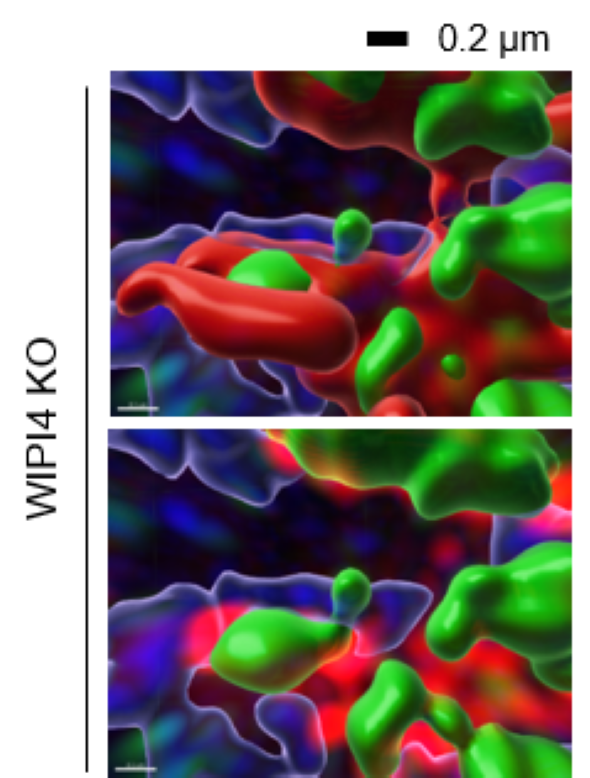
Image: Structured illumination superresolution microscopy of cells where WIPI4 was depleted, showing ATG2A (green) bridging mitochondria (red) and the endoplasmic reticulum (blue)