
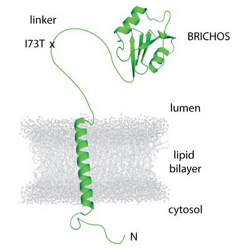
Pulmonary fibrosis (lung scarring) is a serious feature of many different lung diseases, but its precise causes are not well understood. Dr Jennifer Dickens and colleagues from Prof. Stefan Marciniak’s lab are looking to rare, inherited forms of idiopathic pulmonary fibrosis for new insights. Publishing in the European Respiratory Journal, they demonstrate that a pathogenic mutation of surfactant protein C (SFTPC-I73T) shows a toxic gain of function through cellular mislocalisation in alveolar epithelial cells. Normal SFTPC traffics along a multistep path from Golgi to plasma membrane before undergoing endocytosis and final processing within multi-vesicular bodies. In contrast, SFTPC-I73T accumulates in an unprocessed form, cycling between the cell surface and early endosomes.