
The endoplasmic reticulum (ER) constitutes the major cellular compartment for the synthesis, folding, and quality control of secretory proteins. An imbalance between synthesis and folding can lead to ER stress, potentially resulting in ER dysfunction and pathological conditions. To restore cellular homeostasis, adaptive pathways, collectively known as the unfolded protein response (UPR), are activated. The UPR is coordinated by three known ER stress transducers: inositol-requiring kinase 1 (IRE1), protein kinase R-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 alpha (ATF6α). Of the three arms of the UPR, the mechanisms that govern ER stress-dependent ATF6 activation remain the least well understood, even though ATF6 mediates much of the ER-stress-induced changes in gene expression observed in vertebrates.
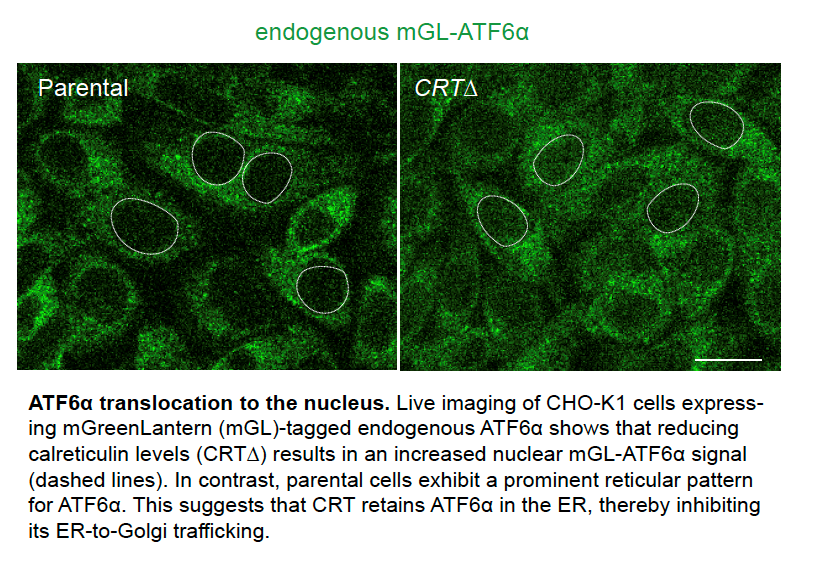
In this new paper led by Adriana Ordoñez from the Ron lab, an ATF6α/IRE1 dual UPR reporter CHO-K1 cell line was generated, and an unbiased genome-wide CRISPR/Cas9 mutagenesis screen was performed. This enabled researchers to systematically profile genetic factors that specifically contribute to ATF6α signalling in the presence and absence of ER stress. The screen identified both anticipated and new candidate genes that regulate ATF6α activation. Among these, calreticulin (CRT), a key ER luminal chaperone, selectively repressed ATF6α signalling. CRT depletion exposed a negative feedback loop implicating ATF6α in repressing IRE1 activity basally and overexpression of CRT reversed this repression. These findings indicate that CRT, beyond its known role as a chaperone, also serves as an ER repressor of ATF6α to selectively regulate one arm of the UPR.