
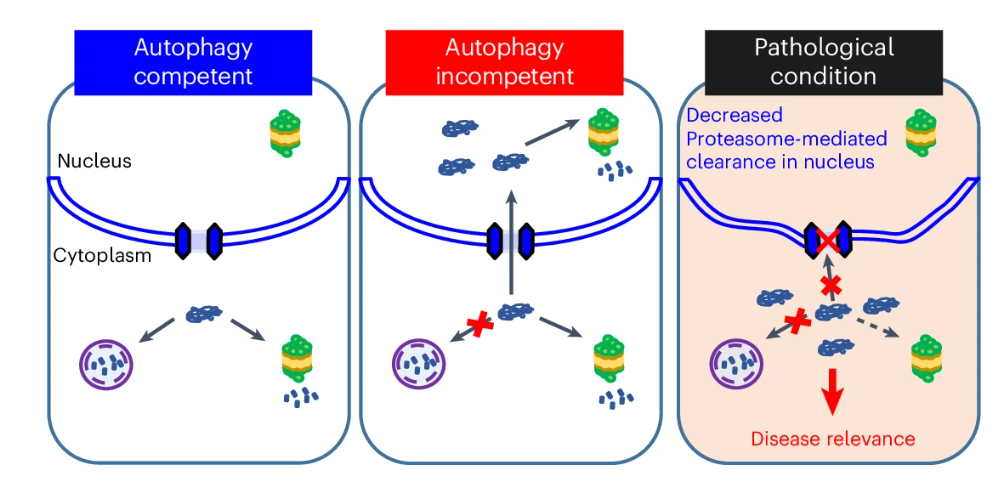
Autophagy is a pathway conserved from yeast to humans enabling the degradation of cytoplasmic proteins and organelles. After such substrates are captured by autophagosomes, these are trafficked to lysosomes where their contents are degraded. Mutations in core autophagy genes cause neurological conditions, with autophagy defects being seen in neurodegenerative diseases like Parkinson’s disease and Huntington’s disease. Given the importance of autophagy in health and disease, it is crucial to understand the processes that are buffered by normal autophagy and the cellular pathway perturbations that autophagy-perturbed cells are vulnerable to. In a new paper in Nature Cell Biology, So Jung Park and colleagues in the Rubinsztein lab have sought to understand this by seeking negative genetic interactions like synthetic lethality in autophagy-null human cells using available data from yeast screens as a starting point. Their studies revealed that loss of proteasome and nuclear pore complex components causes synergistic viability changes akin to synthetic fitness loss in autophagy-null human cells. This can be attributed to the cytoplasm-to-nuclear transport of proteins during autophagy deficiency, and subsequent degradation of these erstwhile cytoplasmic proteins by nuclear proteasomes. As both autophagy and cytoplasm-to-nuclear transport are defective in Huntington’s disease, such cells are more vulnerable to perturbations of proteostasis due to these synthetic interactions. It is proposed that the negative genetic interaction resulting from the loss of these processes may be a much more potent driver of pathology than the multiplicative effects of both modest defects.