
There have been some major developments in computational tools to predict the 3D structures of proteins. Nature recently published the details of AlphaFold2, the machine-learning product of Google DeepMind which created much interest when first announced last November.
A recent publication in Science led by the University of Washington, but with international co-authors including CIMR’s Prof. Randy Read, described RoseTTAFold, another such method. Importantly, both tools are freely accessible to the wider research community, which will increase their value in solving protein structures and generating biological hypotheses, and will also drive greater understanding and faster development of their application and use.
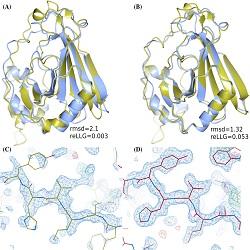
Prof. Read’s group is particularly interested in applying computational tools to molecular replacement, a commonly-used approach to determine protein structures by X-ray crystallography. Their recent paper from Dr Claudia Millán, together with collaborators at the Science and Facilities Technology Council, Max Planck Institute (Tübingen) and University of Liverpool has just been published in Proteins. The authors proposed a new metric (relative expected log-likelihood gain, or reLLG) to evaluate the quality of models for use in molecular replacement – and by which AlphaFold2 performed very favourably. These findings are already assisting in solving particularly difficult protein structures, accelerating understanding of biological and disease processes.